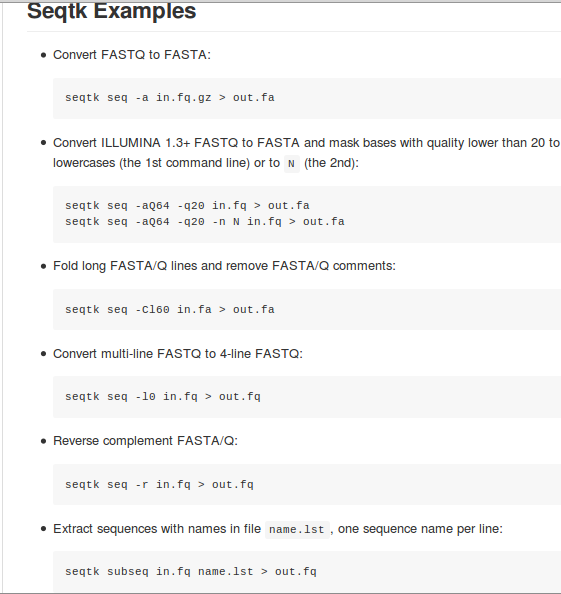
Currently, seqtk supports quality based trimming with the phred algorithm, converting fastq to fasta, reverse complementing sequences, extracting or masking subsequences in regions given in a BED/name list file, and more. It contains a subsampling module to sample exactly n sequences or a fraction of sequences.
Seqtk supports both fasta and fastq input files, which can be optionally gzip compressed.